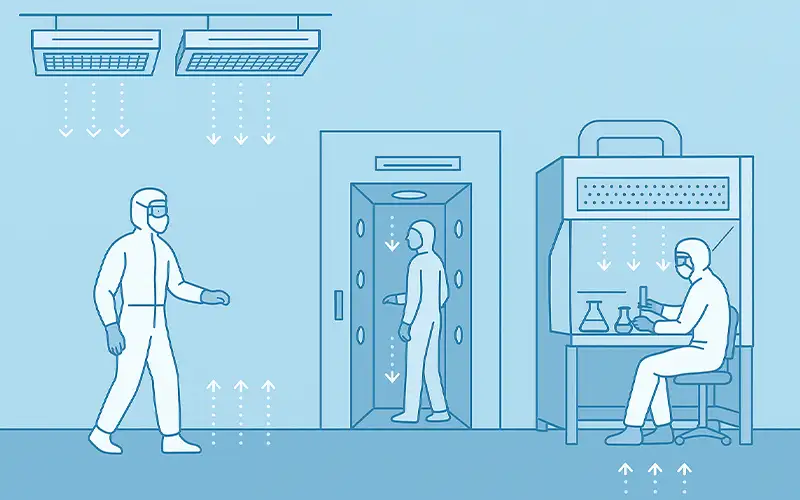
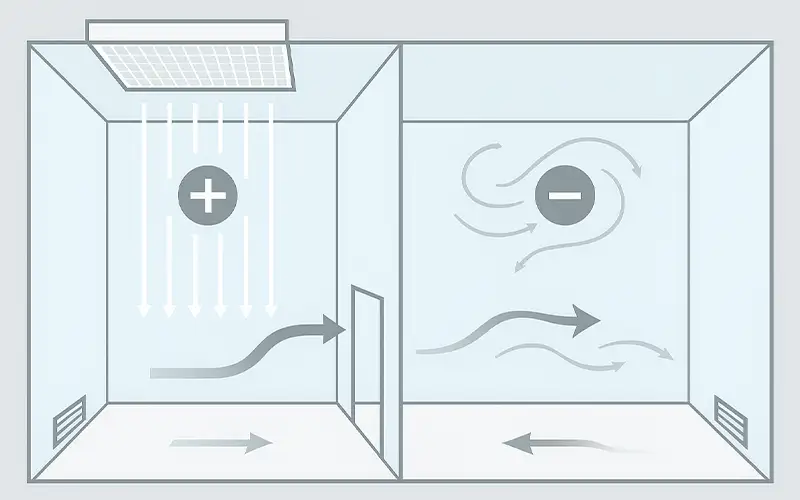
Luftfiltrationsprodukte
Entdecken Sie unser umfassendes Sortiment an hochwertigen Luftfiltrationslösungen, die für verschiedene Branchen konzipiert wurden.
Von HVAC-Filtern über HEPA-Filter bis hin zu industriellen Anwendungen bieten unsere Luftfilter herausragende Leistung, Energieeffizienz und lang anhaltende Zuverlässigkeit.
Finden Sie den perfekten Filter, der Ihren Anforderungen entspricht.